大阪市立大学 ![]() ・大学院理学研究科の杉﨑 研司(すぎさき けんじ)特任講師、佐藤 和信(さとう かずのぶ)、教授、工位 武治(たくい たけじ)名誉教授らの研究チームは12月24日、量子コンピュータを用いてスピン量子数が異なる電子状態(スピン状態)間のエネルギー差を直接計算することができる新規量子アルゴリズム
・大学院理学研究科の杉﨑 研司(すぎさき けんじ)特任講師、佐藤 和信(さとう かずのぶ)、教授、工位 武治(たくい たけじ)名誉教授らの研究チームは12月24日、量子コンピュータを用いてスピン量子数が異なる電子状態(スピン状態)間のエネルギー差を直接計算することができる新規量子アルゴリズム ![]() を発表した。
を発表した。
その要旨は以下通り
(1)量子コンピュータを用いると、原子・分子のエネルギーを精密に求める量子化学計算が超高速で実行できる。
(2)分子は「スピン量子数」が異なると化学反応性なども異なるため、エネルギーが最も低い基底状態のスピン量子数を決定すること、および「スピン量子数」が異なる電子状態(スピン状態)間のエネルギー差を正確に求めることは極めて重要である。
(3)これまで、スピン状態間のエネルギー差を求めるには、それぞれのスピン状態の全エネルギーを計算する必要があった。
(4)量子力学の根本原理である「量子重ね合わせ状態」を上手に利用し、ベイズ推定による機械学習と組み合わせることにより、従来法よりも容易に量子コンピュータに実装でき、スピン状態間のエネルギー差を直接計算できる新規量子アルゴリズムの開発に成功した。
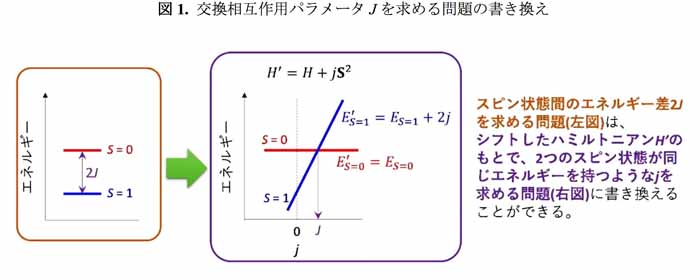
これまで、スピン状態間のエネルギー差を求めるには、たとえ量子コンピュータを用いたとしても、異なるスピン状態の全エネルギーをそれぞれ求める必要があったため、同新規量子アルゴリズムを用いた手法は過去の量子化学計算の常識を覆す画期的なものだ。
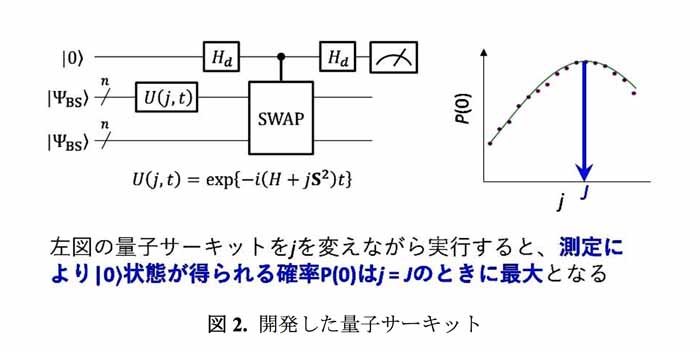
加えて従来手法に於ける〝量子位相推定〟と同様に、計算時間の指数関数的加速が保証されている。かつ量子位相推定よりも量子コンピュータへの実装が容易であること。さらにエラー耐性にも優れた手法でもある。
またそもそも、ほとんどの化学の問題は、系の全エネルギーではなくエネルギー差を議論するものだが、分子サイズが大きくなるにつれて全エネルギーが大きくなるのに対し、議論したいエネルギー差の大きさは分子サイズによらずほぼ一定である。従ってエネルギー差を直接計算することにより、計算量 の分子サイズ依存性を大幅に改善することができ、特に大きな実在分子系の量子化学計算を量子コンピュータで実行するための有力なツールとなるだろう。
新量子アルゴリズムは、量子サーキットの入力として broken- symmetry波動関数以外の波動関数を用いれば、他の問題に簡単に応用することができることから今後、同様の量子サーキットを用いた様々な量子アルゴリズム開発の出発点になるという期待もある。なお同研究成果は、国際学術誌「Chemical Science」(オープンアクセス)に、2020年12月24日(日本時間)掲載される。